Статьи QuantRise
Белки под микроскопом: как точно предсказать их поведение
Автор: Денис Аветисян
Новое исследование демонстрирует, что машинное обучение позволяет значительно повысить точность моделирования стабильности и колебаний белков в различных условиях.
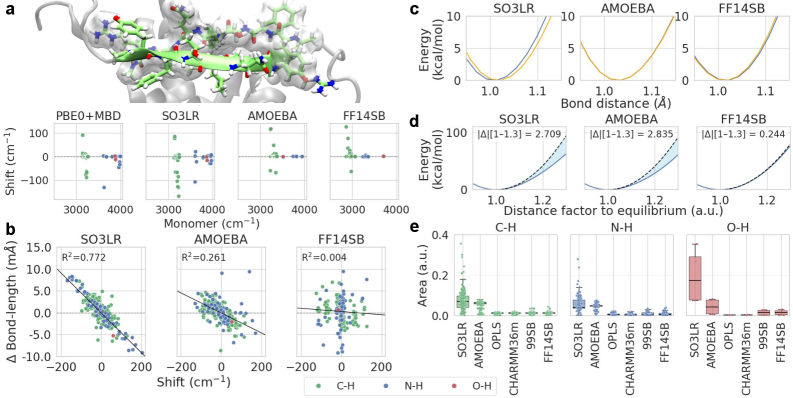
![Исследование вибрационных спектров двадцати трех молекул, включая аспартам, этанол и толуол, демонстрирует, что разработанный метод SO3LR превосходит традиционный GAFF2 в точном воспроизведении результатов расчётов <span class="katex-eq" data-katex-display="false">PBE0+MBD</span>, подтверждённом анализом корреляционных матриц собственных векторов колебаний и сравнением инфракрасных спектров с экспериментальными данными, полученными из баз данных [HITRAN2016\_XSC, HITRAN2020, NISTWebBook2025, Aspartame], что указывает на улучшенное моделирование молекулярных колебаний и потенциальную применимость в более точных расчётах молекулярных свойств.](https://arxiv.org/html/2601.09845v1/x1.png)
Применение машинных силовых полей, обученных на квантово-механических данных, обеспечивает более реалистичные и надежные биомолекулярные симуляции.
Несмотря на значительный прогресс в моделировании биомолекул, точное предсказание их стабильности и колебательных свойств остается сложной задачей. В работе ‘Stability and Vibrations of Proteins in Vacuum and Water: Bridging Quantum Accuracy and Force-Field Efficiency’ представлен новый машинный подход к построению силовых полей (SO3LR), демонстрирующий беспрецедентную точность воспроизведения результатов квантово-химических расчетов для широкого спектра биомолекул. Показано, что SO3LR не только адекватно описывает гармонические и ангармонические колебания, но и позволяет моделировать сложные взаимодействия, определяющие конформацию белков в различных средах. Открывает ли это новые перспективы для создания более надежных и эффективных методов вычислительной биологии и разработки лекарств?
Преодолевая ограничения гармонического приближения: к реалистичным поверхностям потенциальной энергии
Традиционное моделирование молекулярных систем часто опирается на гармоническое приближение, упрощающее поверхность потенциальной энергии (ППЭ). В рамках этого подхода, колебания атомов в молекуле рассматриваются как гармонические, что существенно снижает вычислительные затраты. Однако, реальные молекулярные колебания нелинейны и характеризуются ангармоничностью — отклонением от идеальной гармонической формы. Игнорирование ангармоничности приводит к упрощению ППЭ и, как следствие, к неточностям в предсказании свойств молекул, включая их спектры, скорости реакций и конформационные переходы. В то время как гармоническое приближение может быть полезно для качественного понимания, для получения точных результатов и адекватного описания сложных молекулярных процессов необходимо учитывать ангармонические эффекты и использовать более сложные методы моделирования.
Упрощение, вносимое гармоническим приближением, существенно ограничивает точность предсказания поведения молекулярных систем, особенно в случаях, когда динамика процессов сложна или чувствительна к незначительным изменениям энергии. В реальности, потенциальная энергия молекул не описывается идеально параболической формой, а имеет более сложную, многомерную поверхность. Игнорирование этих отклонений от гармоничности приводит к неточным результатам при моделировании таких явлений, как колебательные спектры, химические реакции и даже процессы, происходящие в биологических макромолекулах, где даже небольшие энергетические различия могут кардинально изменить поведение системы. Следовательно, для получения достоверных предсказаний необходимо учитывать все нюансы формы потенциальной поверхности, включая ее анигармонические компоненты.
Точное описание полной поверхности потенциальной энергии, включая ангармоничность, является ключевым для понимания широкого спектра явлений в химии и биологии. Например, процесс сворачивания белков, определяющий их функциональность, критически зависит от точной формы потенциальной энергии, поскольку даже незначительные изменения в этой поверхности могут радикально повлиять на конформацию белка. Аналогично, анализ колебательных спектров молекул требует учета ангармоничности, поскольку гармоническое приближение не способно адекватно описать реальные частоты и интенсивности линий поглощения. Наконец, понимание механизмов химических реакций, включая скорости и селективность, невозможно без детального знания потенциальной энергии вдоль реакционной координаты, где ангармонические эффекты играют определяющую роль в преодолении энергетического барьера и формировании продуктов реакции.
Современные методы моделирования молекулярных систем сталкиваются со значительной проблемой: необходимо найти баланс между вычислительными затратами и точностью представления потенциальной энергии. Разработка реалистичных поверхностей потенциальной энергии, учитывающих все нюансы молекулярных взаимодействий, требует огромных ресурсов. Упрощения, необходимые для снижения вычислительной нагрузки, часто приводят к потере важной информации о молекулярном движении и реакционной способности. В результате, существующие подходы часто оказываются недостаточно точными для предсказания сложных явлений, таких как процессы сгибания белков или детали вибрационных спектров, требуя постоянного поиска компромиссов и инновационных алгоритмов для эффективного моделирования.
Квантово-механические основы и передовые вычислительные методы
Для получения точных расчетов потенциальных энергетических поверхностей (ПЭП) необходимо учитывать квантово-механические эффекты, такие как поляризация и перенос заряда. Поляризация описывает изменение электронной плотности в молекуле под влиянием внешнего электрического поля или присутствия других молекул, что влияет на межмолекулярные взаимодействия и энергию системы. Перенос заряда возникает при частичном переносе электронной плотности между молекулами, приводя к образованию частично ионного характера связи и модификации электронной структуры. Игнорирование этих эффектов может привести к значительным погрешностям в расчетах энергии, геометрии и других свойств молекул, особенно в системах с выраженными электростатическими взаимодействиями или при рассмотрении химических реакций, включающих перенос электронов.
Методы теории функционала плотности (DFT), такие как PBE0+MBD, представляют собой эффективный подход к учету эффектов поляризации и переноса заряда при расчете поверхностей потенциальной энергии. PBE0 является функционалом обмена-корреляции, улучшающим стандартные функционалы, а MBD (Many-Body Dispersion) — метод, позволяющий корректно описывать дисперсионные взаимодействия, важные для точного моделирования межмолекулярных взаимодействий. Несмотря на свою мощность и точность, DFT-расчеты с такими методами остаются вычислительно затратными, особенно для больших молекулярных систем или при необходимости исследования большого количества конфигураций, что ограничивает их применение в задачах, требующих высокой скорости расчета или масштабного моделирования.
Машинно-обученные силовые поля (MLFF), такие как SO3LR, представляют собой перспективный подход к ускорению исследования поверхностей потенциальной энергии (ППЭ) за счет обучения на данных, полученных с использованием высокоточных квантово-механических расчетов. В частности, SO3LR демонстрирует среднюю абсолютную погрешность при расчете колебательных частот в 12 см⁻¹, что значительно точнее, чем 44 см⁻¹ для традиционного силового поля GAFF2. Это повышение точности позволяет проводить более эффективное моделирование динамики молекул и химических реакций, требующих детального описания колебательных спектров.
Применение современных квантово-механических методов, таких как функционал теории плотности (DFT) с учетом дисперсионных взаимодействий и машинное обучение потенциалов межмолекулярного взаимодействия (MLFF), позволяет получить более точное представление о поверхности потенциальной энергии (ППЭ) по сравнению с традиционными подходами. Традиционные методы часто упрощают описание электронных эффектов, что приводит к искажению формы ППЭ и, следовательно, к неточностям в расчетах свойств молекул и материалов. В частности, MLFF, обученные на данных, полученных высокоточными квантово-механическими расчетами, демонстрируют значительное улучшение точности, например, средняя абсолютная ошибка для колебательных частот составляет около 12 cm^{-1} по сравнению с 44 cm^{-1} для GAFF2, что позволяет более реалистично моделировать динамику молекулярных систем.
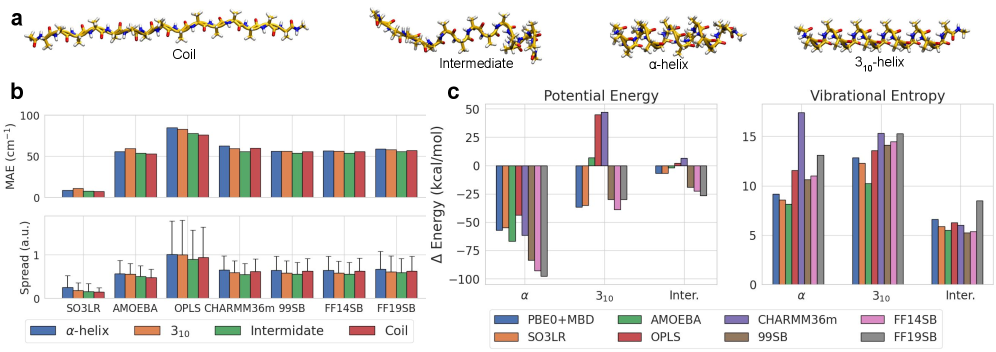
Моделирование молекулярной динамики и колебательных спектров
Молекулярная динамика (МД) представляет собой вычислительный метод, позволяющий отслеживать изменение во времени структуры и свойств молекулярных систем. В основе МД лежит численное решение уравнений движения для каждого атома в системе, используя классическую механику. Для вычисления сил, действующих на атомы, используются силовые поля, такие как GAFF2 или более современные машинные обучаемые силовые поля (MLFF), например SO3LR. Эти силовые поля описывают потенциальную энергию системы в зависимости от координат атомов, позволяя рассчитывать силы и, следовательно, траектории движения атомов во времени. Результатом МД-симуляции является временная эволюция системы, которая может быть использована для анализа различных свойств, включая структурные изменения, динамические характеристики и термодинамические свойства.
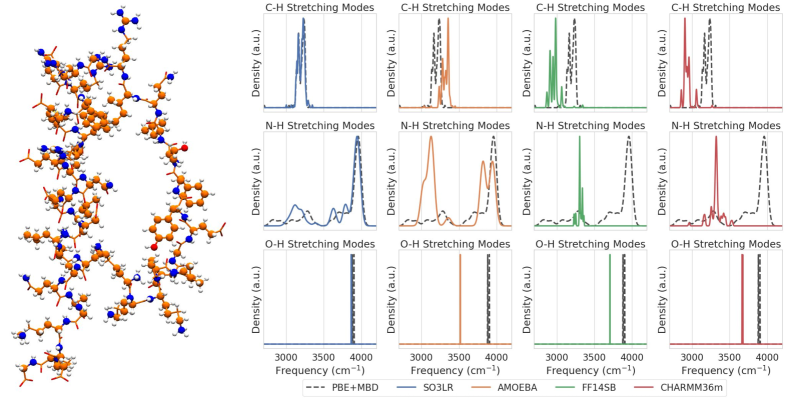
Комбинирование молекулярной динамики (МД) с нормальным модным анализом (НМА) позволяет предсказывать частоты колебаний и инфракрасные спектры молекул. Методы, такие как PBE0+MBD или SO3LR, используются для расчета гессиана — матрицы вторых производных энергии по координатам, необходимой для определения нормальных мод. МД используется для получения ансамбля молекулярных конфигураций, а НМА применяется к каждой конфигурации для вычисления частот колебаний. Полученные спектры можно сравнивать с экспериментальными данными, полученными с помощью инфракрасной спектроскопии, для валидации модели и изучения динамических свойств молекулярных систем. Важно отметить, что точность предсказания частот напрямую зависит от качества используемого функционала плотности (например, PBE0) и метода учета дисперсионных взаимодействий (например, MBD).
Молекулярные динамические симуляции позволяют исследовать сложные процессы, такие как сворачивание белков и конформационные изменения. В качестве модельного объекта для изучения сворачивания часто используется пептид AceAla15NMe, что позволяет анализировать динамику изменения конформации и стабильности структуры. Эти симуляции дают возможность отслеживать временную эволюцию молекулярной системы, выявлять ключевые этапы процесса сворачивания и исследовать факторы, влияющие на стабильность конечной конформации. Анализ динамики конформационных изменений позволяет понять механизмы функционирования молекул и предсказывать их поведение в различных условиях.
Точность моделирования молекулярной динамики и предсказания колебательных спектров напрямую зависит от качества потенциальной энергетической поверхности (ПЭС) и выбранного силового поля. В частности, силовое поле SO3LR демонстрирует более тесную корреляцию между изменениями длин связей и сдвигами в колебательных частотах по сравнению с многими традиционными силовыми полями, такими как GAFF2. Это означает, что SO3LR более адекватно отражает влияние изменений геометрии молекулы на её колебательные характеристики, что критически важно для точного предсказания инфракрасных спектров и анализа динамического поведения молекул.
Приложения и последствия для биологических систем
Точное моделирование потенциальной энергии (ПЭС) и молекулярной динамики (МД) являются основополагающими инструментами для раскрытия динамики биологических молекул, таких как белки и нуклеиновые кислоты. Эти вычислительные методы позволяют исследователям не просто статично изобразить структуру, но и наблюдать, как молекулы изменяют свою форму и взаимодействуют во времени. Моделирование ПЭС, основанное на квантово-механических расчетах, определяет энергетические ландшафты, в которых “плавают” молекулы, а МД-симуляции отслеживают их движение по этим ландшафтам, учитывая влияние температуры и растворителя. Благодаря этому подходу можно исследовать конформационные изменения, необходимые для выполнения биологических функций, предсказывать стабильность молекул и выявлять ключевые взаимодействия, определяющие их поведение. Такая точность имеет решающее значение для понимания сложных биологических процессов на молекулярном уровне и разработки новых терапевтических стратегий.
Использование модифицированных аминокислот, таких как L-ooF-фенилаланин+H+, открывает новые возможности для исследования конформационных ландшафтов биомолекул. Вводя неестественные аминокислоты в структуру белка, исследователи получают инструменты для более детального изучения энергетических минимумов и, что особенно важно, метастабильных состояний, которые могут играть ключевую роль в функционировании белка. Данный подход позволяет не только картировать энергетические поверхности, но и выявлять промежуточные конформации, невидимые при изучении исключительно природных белков. Это, в свою очередь, способствует более глубокому пониманию механизмов сворачивания белка, его взаимодействия с другими молекулами и, в конечном итоге, его биологической функции. Анализ метастабильных состояний с использованием модифицированных аминокислот представляет собой перспективный путь для разработки новых лекарственных препаратов и создания материалов с заданными свойствами.
Разработанные методы моделирования потенциальных энергетических поверхностей и молекулярной динамики открывают значительные перспективы в различных областях науки. В частности, они представляют собой мощный инструмент для разработки новых лекарственных препаратов, позволяя моделировать взаимодействие молекул лекарства с биологическими мишенями и оптимизировать их структуру для повышения эффективности и селективности. В материаловедении эти методы способствуют созданию материалов с заданными свойствами, предсказывая их поведение на молекулярном уровне. Особый интерес представляет возможность углубленного изучения сложных биомолекул, таких как белок p53, играющий ключевую роль в регуляции клеточного цикла и развитии рака. Точное моделирование конформационных изменений и динамики p53 позволяет понять механизмы функционирования и разработать стратегии для восстановления его активности в раковых клетках, что потенциально открывает новые пути для терапии онкологических заболеваний.
Понимание дипольных моментов и поляризуемости имеет решающее значение для корректной интерпретации инфракрасной спектроскопии и прогнозирования межмолекулярных взаимодействий. Исследования показали, что модифицированная молекула SO3LR демонстрирует стабильно низкие показатели разброса при анализе ее колебательных мод, что указывает на улучшенное соответствие между теоретическими расчетами и экспериментальными данными. Это свидетельствует о более точном моделировании колебательных свойств молекулы и, как следствие, о повышенной надежности предсказаний относительно ее поведения в биологических системах и способности к взаимодействию с другими молекулами. Полученные результаты позволяют более эффективно использовать инфракрасную спектроскопию для изучения структуры и динамики биомолекул, а также разрабатывать новые подходы к созданию материалов с заданными свойствами.
Исследование демонстрирует, что точность моделирования биомолекулярных систем напрямую зависит от адекватности используемых силовых полей. Разработанное в работе силовое поле SO3LR, обученное на квантово-механических данных, превосходит традиционные аналоги в предсказании стабильности и колебаний белков. Это согласуется с глубоким пониманием системного подхода, когда поведение сложной структуры определяется не только её компонентами, но и взаимодействием между ними. Как однажды заметил Пётр Капица: «В природе нет ничего случайного, все подчинено строгим законам». В данном исследовании эти законы находят своё отражение в точности моделирования, позволяя предсказывать поведение биомолекул с беспрецедентной степенью достоверности.
Куда Далее?
Представленная работа, безусловно, демонстрирует прогресс в моделировании биомолекулярной стабильности и колебаний. Однако, эйфория от повышения точности, достигаемого машинным обучением, должна быть умеренной. Иначе рискуем оптимизировать не то, что необходимо, а лишь то, что легко измерить. Улучшение силы поля — это, конечно, важно, но истинный вызов заключается в понимании и моделировании сложных взаимосвязей между структурой и динамикой в реальных биологических системах.
Простота масштабируется, изощрённость — нет. SO3LR, несомненно, представляет собой шаг вперёд, но зависимость от квантово-химических данных остаётся настоящей ценой свободы. Необходимо сосредоточиться на разработке методов, позволяющих уменьшить эту зависимость, создавая самосогласованные модели, способные к адаптации и самообучению. Хорошая архитектура незаметна, пока не ломается, и нам следует стремиться к такой же элегантности в наших моделях.
В конечном итоге, подлинный прогресс требует не только повышения точности, но и развития принципиально новых подходов к моделированию. Необходимо учитывать эффекты, которые сейчас кажутся незначительными, но могут оказаться критически важными для понимания сложных биологических процессов. И, возможно, нам следует признать, что некоторые проблемы просто не поддаются решению с использованием существующих методов.
Оригинал статьи: https://arxiv.org/pdf/2601.09845.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Статья также опубликована на личном сайте автора.