Квантовые технологии
Квантовая химия: от теории к практике
Автор: Денис Аветисян
- Вызов точности: Фундаментальные трудности в энергетическом моделировании молекул
- Sample-based Quantum Diagonalization (SQD): Новый горизонт в расчете электронной структуры
- Уточнение точности: Методы смягчения ошибок и экстраполяции в SQD
- Валидация и оценка: Тестирование SQD на наборе данных W4-11
- Куда Ведет Этот Путь?
Новое исследование сравнивает эффективность квантовых алгоритмов с классическими методами для предсказания термохимических свойств молекул.
Проведена всесторонняя оценка метода Sample-Based Quantum Diagonalization на наборе данных W4-11, демонстрирующая необходимость экстраполяции для достижения точности, сравнимой с традиционными подходами.
Несмотря на теоретический потенциал квантовых вычислений в моделировании молекулярных систем, практическая реализация и оценка эффективности квантовых алгоритмов остаются сложной задачей. В работе ‘From Promise to Practice: Benchmarking Quantum Chemistry on Quantum Hardware’ представлен всесторонний сравнительный анализ метода Sample-Based Quantum Diagonalization (SQD) для расчета термохимических свойств, основанный на широко известном наборе данных W4-11. Полученные результаты демонстрируют, что, несмотря на значительные статистические отклонения, использование методов экстраполяции позволяет достичь точности, сопоставимой с классическими методами корреляционных вычислений, такими как CCSD. Какие дальнейшие усовершенствования алгоритмов и аппаратного обеспечения необходимы для раскрытия полного потенциала квантовой химии и преодоления существующих ограничений?
Вызов точности: Фундаментальные трудности в энергетическом моделировании молекул
Определение точных энергий молекул является фундаментальной задачей для прогнозирования химических реакций и свойств материалов, однако эта задача сопряжена со значительными вычислительными трудностями. Точность моделирования напрямую влияет на предсказание скорости реакции, стабильности соединений и различных физических характеристик, что делает ее критически важной для многих областей науки и техники. Несмотря на прогресс в вычислительной химии, расчет энергий даже относительно небольших молекул может потребовать огромных ресурсов и времени, ограничивая возможности изучения сложных систем, таких как белки или полимеры. Поиск эффективных и точных методов расчета для молекул остается одной из ключевых проблем современной теоретической химии и физики.
Традиционные методы квантовой химии, такие как , демонстрируют высокую точность в вычислении энергии молекул, однако их вычислительная сложность резко возрастает с увеличением числа атомов в системе. Этот факт обусловлен необходимостью учета корреляций между электронами, что требует экспоненциального увеличения вычислительных ресурсов. В результате, применение и подобных методов ограничено изучением лишь небольших молекул, состоящих из нескольких десятков атомов. Это создает значительные препятствия для моделирования более сложных систем, представляющих интерес в химии, биологии и материаловедении, и стимулирует поиск альтернативных, масштабируемых подходов к расчету электронной структуры.
Ограничения в вычислительных ресурсах, препятствующие точному определению энергий молекул, оказывают существенное влияние на прогресс в таких областях, как разработка лекарственных препаратов и материаловедение. Поиск новых лекарственных средств требует моделирования взаимодействия миллионов молекул с биологическими мишенями, а создание материалов с заданными свойствами — предсказания их поведения на атомном уровне. Эти задачи, требующие высокой точности расчетов, зачастую оказываются непосильными для традиционных методов, что стимулирует поиск инновационных подходов к вычислению электронной структуры веществ. Разработка эффективных алгоритмов и использование передовых вычислительных технологий становятся ключевыми факторами для преодоления этого препятствия и ускорения научных открытий в химии и физике материалов.
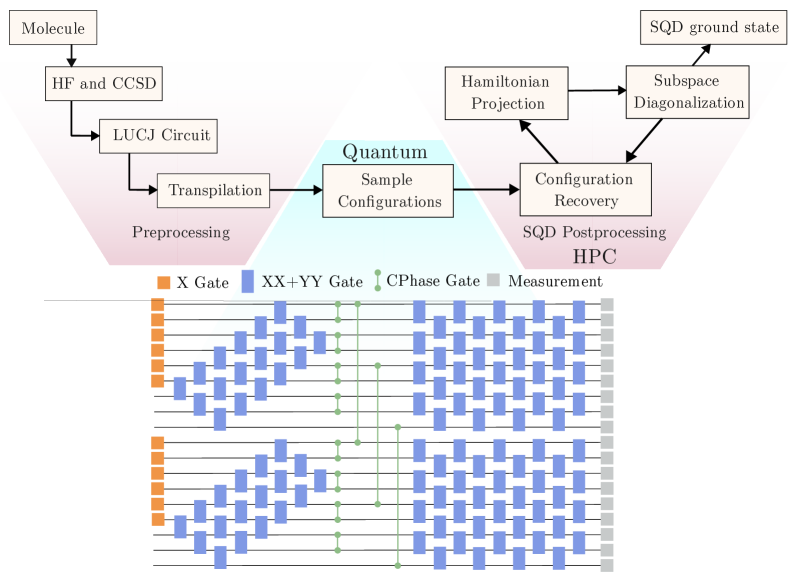
Sample-based Quantum Diagonalization (SQD): Новый горизонт в расчете электронной структуры
Метод Sample-based Quantum Diagonalization (SQD) представляет собой гибридный квантово-классический алгоритм, предназначенный для исследования электронной структуры молекул. В отличие от традиционных методов, требующих экспоненциальных вычислительных ресурсов, SQD использует квантовые схемы для получения выборок (samples) электронной структуры. Этот подход позволяет снизить вычислительную сложность, поскольку вместо полного вычисления волновой функции, алгоритм фокусируется на получении статистически значимых выборок, которые затем обрабатываются классическими вычислениями для определения энергии и других свойств молекулы. Эффективность SQD заключается в возможности аппроксимации волновой функции с использованием относительно небольших квантовых схем, что делает его перспективным для моделирования более крупных и сложных молекул.
В основе алгоритма Sample-based Quantum Diagonalization (SQD) лежит использование вариационного ответа, представленного в виде Local Unitary Coupled Cluster Jastrow (LUCJ) анзаца. LUCJ представляет собой эффективный способ аппроксимации волновой функции молекулы, используя локальные унитарные преобразования, примененные к определенной базовой функции, часто включающей Jastrow корреляционный фактор. Это позволяет существенно снизить вычислительные затраты, связанные с представлением сложных электронных корреляций, необходимых для точного расчета энергии молекулы. В контексте SQD, LUCJ анзац кодируется в квантовую схему, где параметры анзаца оптимизируются вариационным методом для минимизации энергии, полученной из квантовых измерений.
Алгоритм Sample-based Quantum Diagonalization (SQD) сочетает в себе квантическое сэмплирование и классическую постобработку для решения задачи электронных структурных расчетов. Квантическое сэмплирование позволяет эффективно оценить свойства молекулярной системы, снижая вычислительные затраты по сравнению с традиционными методами. Классическая постобработка, включающая анализ полученных квантовых выборок, используется для реконструкции энергетических уровней и других важных параметров молекулы. Такое гибридное сочетание подходов открывает перспективы для масштабирования расчетов на более сложные молекулярные системы, сохраняя при этом приемлемую точность результатов и потенциально преодолевая ограничения, связанные с экспоненциальным ростом вычислительных ресурсов при увеличении размера молекулы.
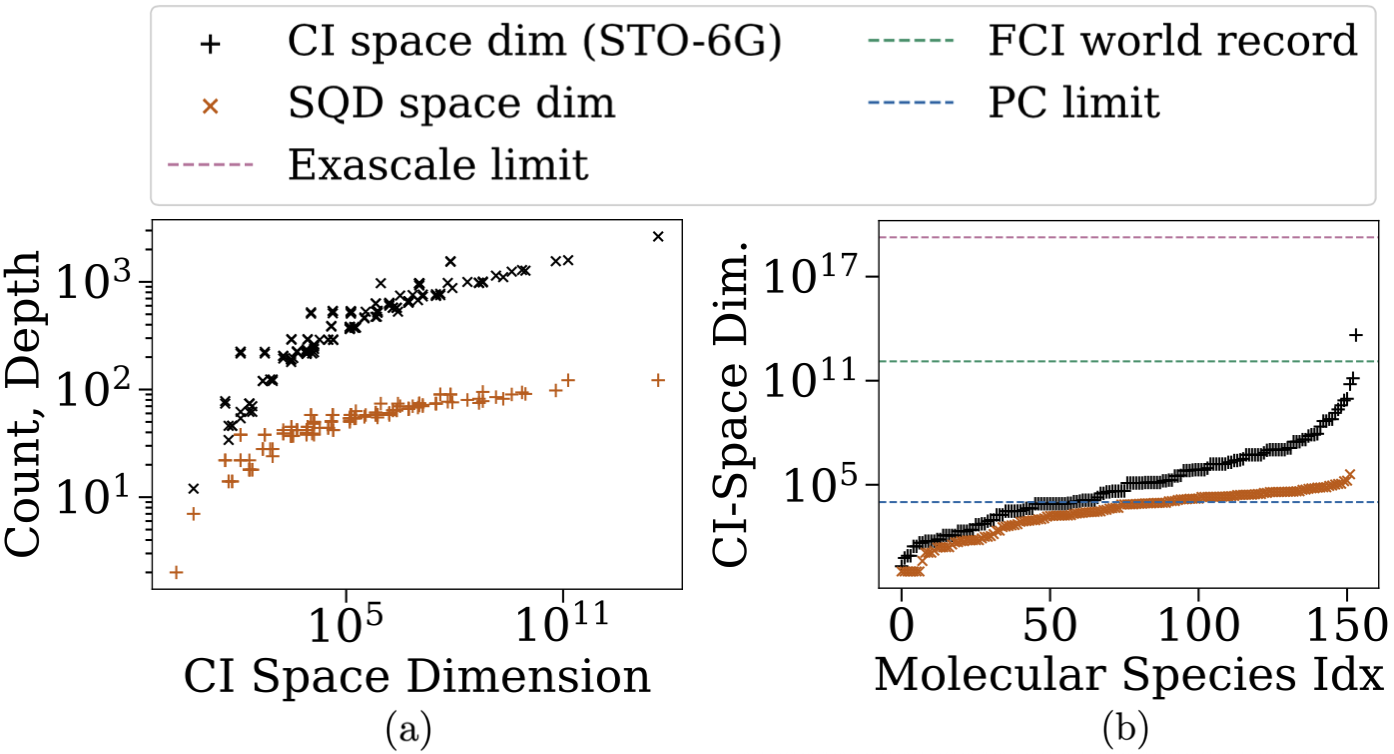
Уточнение точности: Методы смягчения ошибок и экстраполяции в SQD
Методы смягчения ошибок играют критически важную роль в обеспечении достоверности результатов квантовых вычислений, в частности, при использовании спектрального квантового детектирования (SQD). Квантовые системы подвержены различным источникам шума и несовершенств, включая декогеренцию, ошибки управления и шум измерений. Эти факторы приводят к искажению квантовых состояний и, как следствие, к неточностям в вычислениях. Методы смягчения ошибок направлены на подавление или компенсацию этих нежелательных эффектов, позволяя получить более надежные и точные оценки энергетических уровней и других квантовых свойств исследуемой системы. Без применения этих методов, даже небольшие погрешности могут существенно повлиять на достоверность получаемых результатов, особенно в задачах, требующих высокой точности, таких как определение структуры молекул или моделирование квантовых материалов.
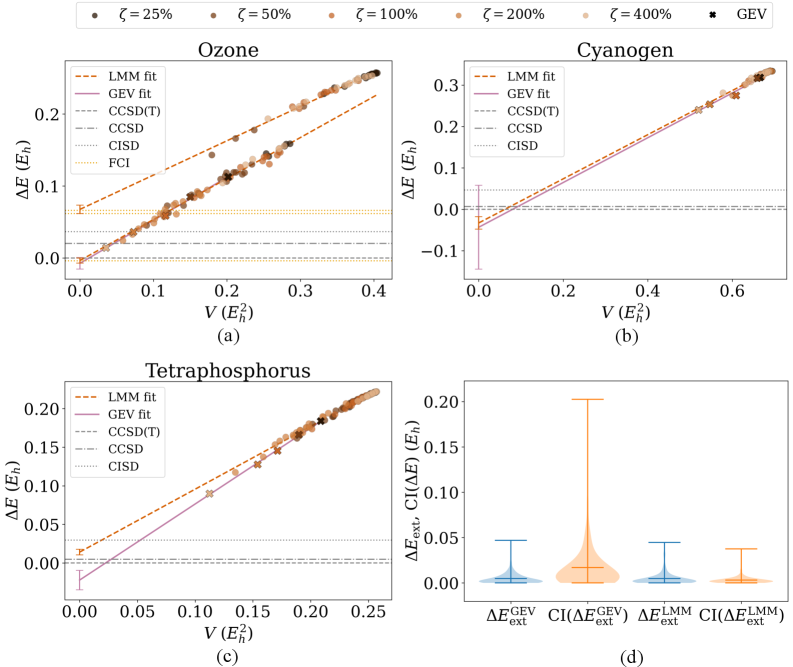
Метод экстраполяции обобщенных собственных значений (Generalized Eigenvalue Extrapolation, GEV Extrapolation) используется для повышения точности вычислений в SQD (Simulated Quantum Dynamics) путем систематической корректировки оценок энергии. В рамках этого метода, энергия, полученная при различных приближениях или параметрах, экстраполируется к пределу, где влияние приближений минимально. Это достигается путем анализа собственных значений — гамильтониана системы — при различных степенях обобщения, что позволяет получить более точное значение энергии основного состояния или других интересующих энергетических уровней. Процедура предполагает построение зависимости между собственными значениями и порядком обобщения, и последующую экстраполяцию этой зависимости для получения уточненной оценки энергии.
Дополнительная оптимизация параметров и повышение скорости сходимости в методе SQD достигается за счет применения модели линейной смеси (LMM) и анализа дисперсии энергии. Модель LMM позволяет аппроксимировать распределение энергий, полученное в результате квантовых вычислений, как комбинацию нескольких линейных компонент, что позволяет более точно оценить истинные энергетические уровни. Анализ дисперсии энергии, в свою очередь, позволяет определить вклад различных источников шума и неопределенности в конечный результат, что позволяет целенаправленно улучшать параметры модели и повышать стабильность сходимости алгоритма. Применение данных методов позволяет минимизировать влияние погрешностей и получить более надежные результаты SQD.
Валидация и оценка: Тестирование SQD на наборе данных W4-11
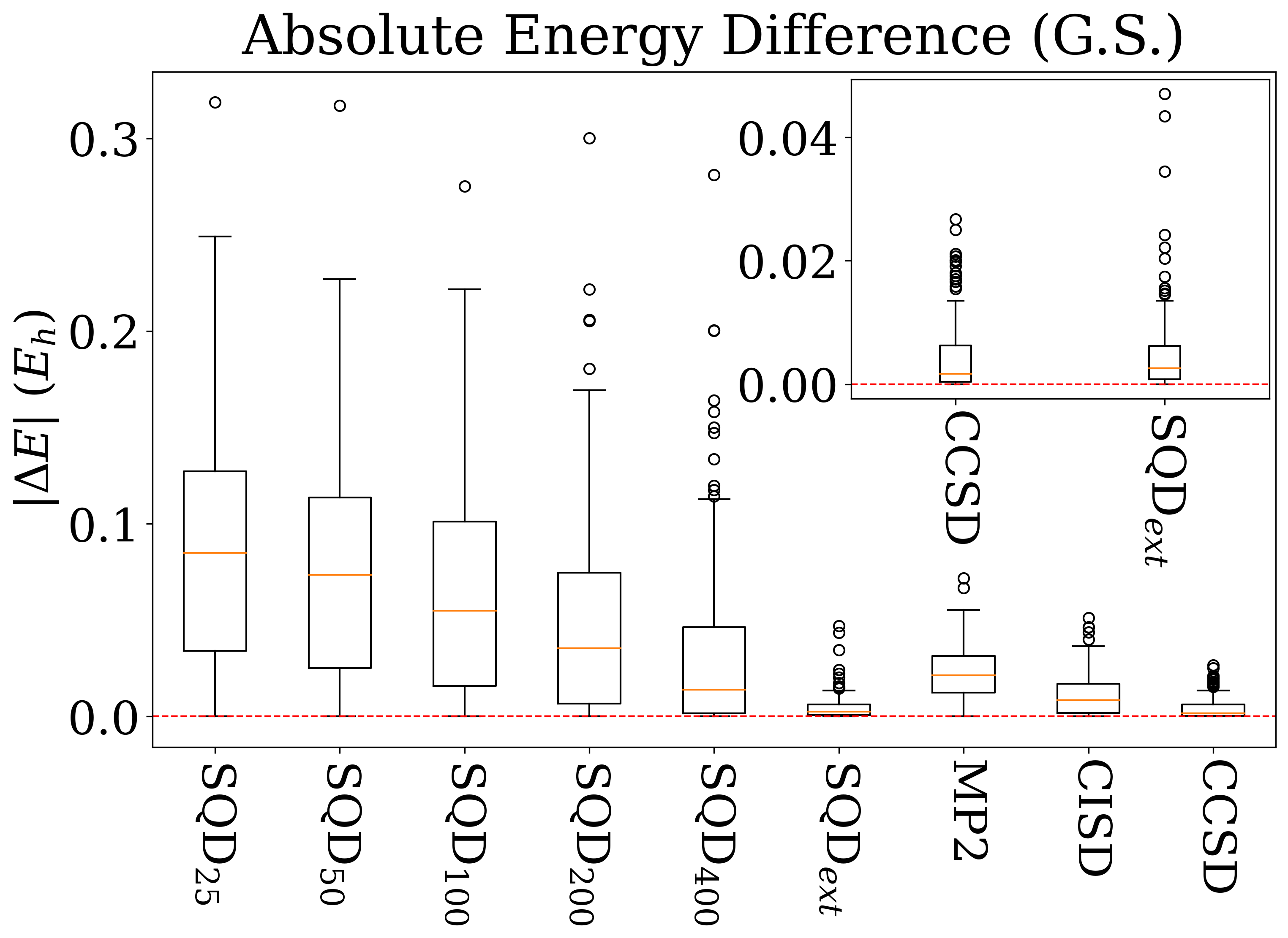
Для всесторонней оценки предложенного метода SQD была проведена строгая проверка на общепризнанном наборе данных W4-11, включающем 745 термохимических реакций, который широко используется для калибровки и валидации методов квантовой химии. Данный набор, охватывающий разнообразные типы реакций и молекулярные системы, позволяет объективно оценить точность и надежность SQD в предсказании энергий реакций и стабильности молекул. Использование W4-11 в качестве эталонного набора гарантирует возможность сопоставления результатов, полученных с помощью SQD, с результатами, полученными с помощью устоявшихся методов, таких как CCSD, что обеспечивает прозрачность и достоверность оценки эффективности нового подхода.
Для снижения вычислительных затрат при проведении термохимических расчетов, в рамках исследования применялся минимальный базисный набор и приближение замороженного ядра. Использование позволяет существенно сократить размер матриц, с которыми необходимо оперировать, а приближение замороженного ядра фиксирует электроны внутренних оболочек, исключая их из рассмотрения в процессе вычислений. Данный подход, несмотря на упрощения, обеспечивает разумную точность результатов, позволяя эффективно оценивать энергии атомизации и диссоциации связей, и является компромиссом между скоростью вычислений и достижением высокой точности.
Результаты тестирования SQD на наборе данных W4-11 демонстрируют его конкурентоспособность по сравнению с традиционными методами, такими как CCSD. Вычисления показали, что SQD обеспечивает сопоставимую точность при расчете (Total Atomization Energy) и (Bond Dissociation Energy), достигая медианной абсолютной ошибки в 0.0030 Эв, что близко к показателю CCSD в 0.0032 Эв. Особого внимания заслуживает метод экстраполяции SQDext, который значительно улучшил межквартильный размах для реакций переноса тяжелых атомов (HAT), снизив его до 0.0277 Эв, и уменьшил максимальные абсолютные ошибки для реакций до 0.0758 Эв. Это свидетельствует о повышенной стабильности и снижении количества выбросов, что делает SQD надежным инструментом для термохимических расчетов.
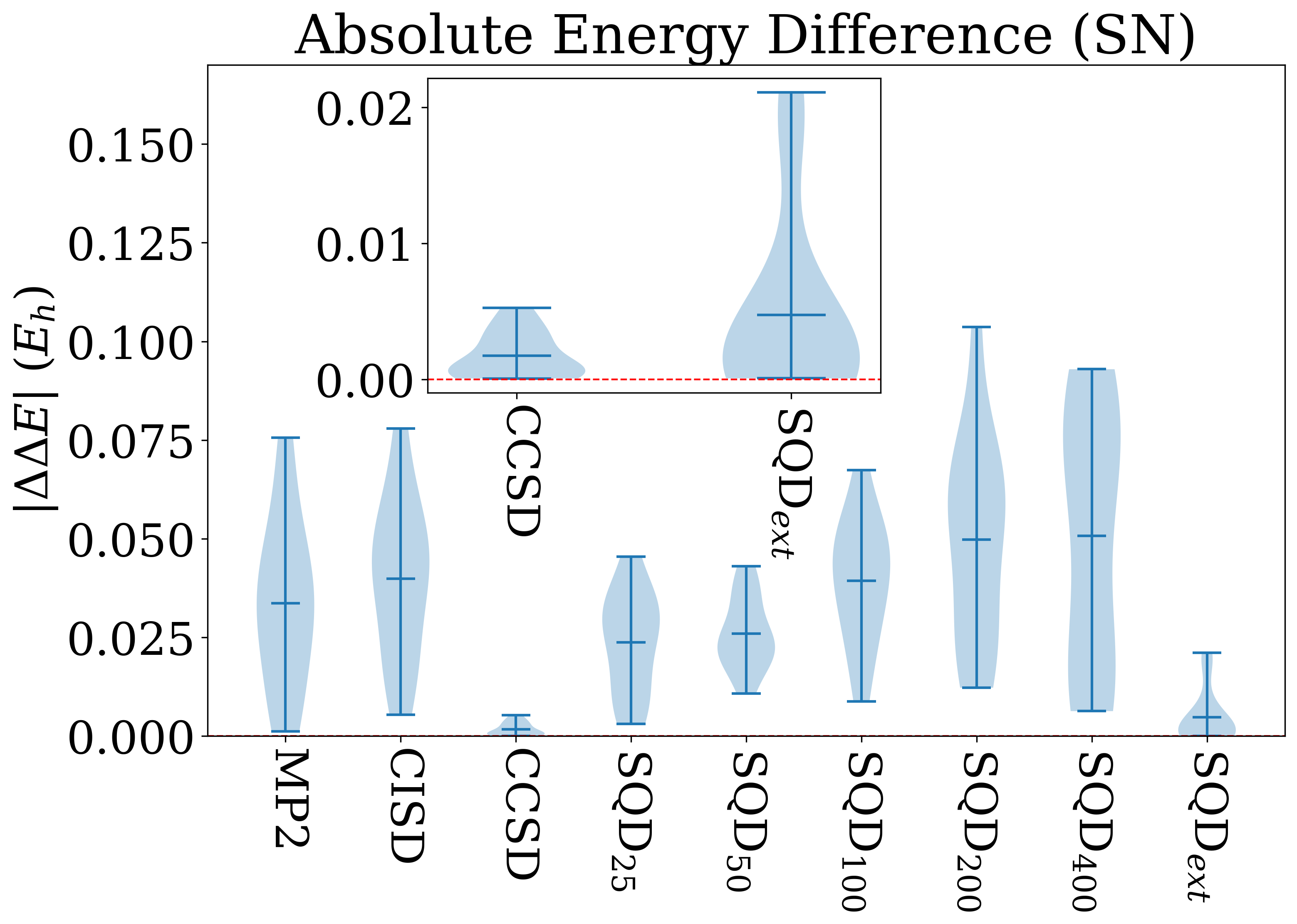
Исследование, представленное в данной работе, демонстрирует, что методы квантовой химии, такие как Sample-Based Quantum Diagonalization (SQD), хоть и перспективны, требуют дальнейшей оптимизации и применения методов экстраполяции для достижения сопоставимой точности с классическими подходами. Подобно тому, как каждая версия программного обеспечения является итерацией к совершенству, SQD нуждается в постоянном улучшении. Как заметил Эрвин Шрёдингер: «Нельзя сказать, что физика описывает реальность, она лишь описывает то, что мы можем измерить». Это отражает суть работы — стремление к более точным измерениям и, следовательно, более глубокому пониманию электронных структур и термохимических свойств, опираясь на данные W4-11.
Куда Ведет Этот Путь?
Представленная работа, подобно любому измерению, лишь очерчивает границы незнания. Метод Sample-Based Quantum Diagonalization (SQD), продемонстрировавший определенные перспективы в предсказании термохимических свойств, все же требует дальнейшей доработки. В частности, необходимость экстраполяционных техник для достижения сопоставимой с классическими методами точности указывает на то, что квантовое преимущество, если оно и существует, пока еще скрыто в слоях вычислительной сложности.
Вместо того, чтобы стремиться к немедленному превосходству над устоявшимися алгоритмами, представляется более продуктивным рассматривать SQD как инструмент для исследования самой природы вычислительной точности. Как и эрозия, «технический долг» в квантовых вычислениях проявляется не сразу, но постепенно подрывает надежность результатов. Поиск методов смягчения этой «эрозии» — вот где кроется подлинный вызов.
В конечном счете, истинная ценность подобных исследований заключается не в достижении конкретных цифр точности, а в понимании того, как системы — будь то молекулы или алгоритмы — стареют во времени. Аптайм — это редкая фаза гармонии во времени, и задача науки — не просто её продлить, но и понять закономерности, управляющие её неизбежным угасанием.
Оригинал статьи: https://arxiv.org/pdf/2512.01012.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Статья также опубликована на личном сайте автора.