Квантовые технологии
Квантовые вычисления с ускорением: новый подход к моделированию молекул
Автор: Денис Аветисян
Исследователи предлагают гибридный квантово-классический алгоритм, сочетающий машинное обучение с учетом физических законов и квантовые вычисления для повышения эффективности и точности расчетов электронной структуры.
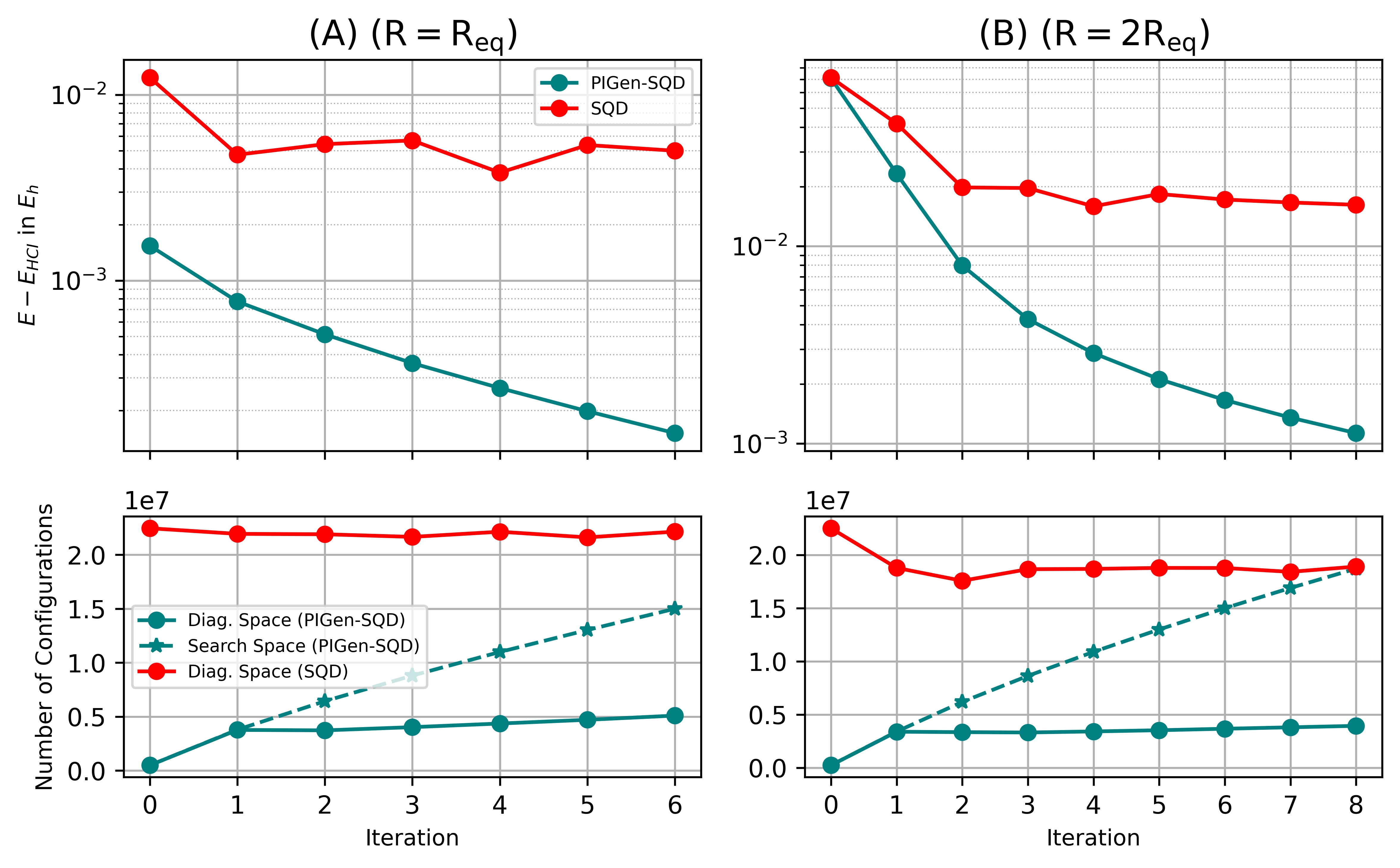
Представлен алгоритм PIGen-SQD, объединяющий машинное обучение, учитывающее физические ограничения, и квантово-центричные супервычисления для ускорения расчетов электронной структуры путем снижения вычислительных затрат на восстановление конфигураций.
Несмотря на значительный прогресс в квантовых вычислениях, достижение практической пользы в решении сложных задач остается сложной задачей. В настоящей работе, посвященной ‘Physics Informed Generative Machine Learning for Accelerated Quantum-centric Supercomputing’, предлагается новый гибридный квантово-классический алгоритм PIGen-SQD, использующий машинное обучение с учетом физических ограничений для ускорения квантово-центрического супервычисления. Данный подход позволяет эффективно восстанавливать доминирующие конфигурации фермионного пространства, снижая вычислительные затраты на диагонализацию гамильтониана и повышая точность расчетов электронных структур. Способствует ли сочетание квантовых вычислений и интеллектуальных алгоритмов созданию надежных и масштабируемых методов для моделирования сложных квантовых систем?
Пределы Классической Квантовой Химии
Точное моделирование молекулярных систем с использованием , или стационарного уравнения Шрёдингера, является фундаментальным требованием в современной химии и физике. Однако, сложность вычислений экспоненциально возрастает с увеличением числа атомов в молекуле. Это связано с тем, что волновая функция, описывающая состояние молекулы, зависит от координат всех электронов, что приводит к огромному объему вычислений даже для относительно небольших молекул. В результате, точное решение становится практически невозможным для сложных систем, таких как белки или большие органические молекулы, что существенно ограничивает возможности исследования их свойств и поведения. Разработка эффективных приближений и вычислительных методов, позволяющих обойти эти ограничения, является одной из ключевых задач современной теоретической химии.
Традиционные методы квантовой химии, демонстрирующие высокую эффективность при моделировании относительно простых молекулярных систем, сталкиваются с серьезными ограничениями при увеличении их размера. Сложность вычислений растет экспоненциально с увеличением числа электронов и атомных ядер, что обусловлено необходимостью решения -частичного уравнения Шрёдингера. Например, для точного описания взаимодействия даже умеренно больших молекул, таких как пептиды или небольшие белки, требуются вычислительные ресурсы, недоступные на современных суперкомпьютерах. Это связано с тем, что количество базисных функций, необходимых для адекватного представления волновой функции, растет экспоненциально, что делает прямые методы непрактичными для систем, содержащих более нескольких десятков атомов. В результате, приходится прибегать к упрощениям и приближениям, которые, хотя и позволяют проводить расчеты, могут существенно повлиять на точность полученных результатов и ограничить возможности моделирования сложных химических процессов.
Приближение Борна-Оппенгеймера, являющееся краеугольным камнем квантово-химических расчетов, значительно упрощает задачу решения уравнения, разделяя движение ядер и электронов. Однако, данное упрощение вносит ограничения при изучении динамических процессов, таких как химические реакции или взаимодействие молекул с электромагнитным излучением. В частности, приближение не учитывает неадиабатические эффекты, возникающие при близком расположении потенциальных поверхностей, что приводит к неточному описанию возбужденных состояний и переходов между ними. Поэтому, хотя приближение Борна-Оппенгеймера остается незаменимым инструментом в большинстве случаев, для адекватного моделирования процессов, где электронно-ядерная корреляция играет существенную роль, требуются более сложные методы, учитывающие взаимодействие между движением электронов и ядер.
Ограничения, с которыми сталкиваются традиционные методы квантохимического моделирования, оказывают существенное влияние на прогресс в различных областях науки и техники. В материаловедении, точность предсказания свойств новых материалов, необходимых для создания инновационных технологий, напрямую зависит от возможности адекватного описания электронного строения сложных систем. В фармацевтической отрасли, разработка эффективных лекарственных препаратов требует детального понимания взаимодействия молекул лекарства с биологическими мишенями, что затрудняется неспособностью существующих методов точно моделировать большие и гибкие молекулы. Наконец, углубление фундаментальных знаний о химических процессах, таких как фотосинтез или катализ, требует преодоления ограничений, связанных с описанием динамических эффектов и возбужденных состояний, что открывает новые возможности для разработки более эффективных и устойчивых технологий. Таким образом, преодоление этих вычислительных барьеров является ключевой задачей для дальнейшего развития химии и смежных наук.
Квантовые Вычисления: Новая Парадигма Молекулярного Моделирования
Квантовые вычисления предлагают принципиально новые возможности для решения задач, недоступных классическим компьютерам, благодаря использованию квантовых явлений суперпозиции и запутанности. Суперпозиция позволяет кубиту представлять собой комбинацию состояний 0 и 1 одновременно, в отличие от классического бита, который может находиться только в одном из этих состояний. Запутанность, в свою очередь, создает корреляцию между кубитами, позволяя им мгновенно влиять друг на друга, независимо от расстояния. Эти свойства обеспечивают экспоненциальный рост вычислительной мощности по мере увеличения количества кубитов, что позволяет моделировать сложные системы, такие как молекулы, с гораздо большей точностью и эффективностью, чем это возможно на классических компьютерах. В частности, для кубитов, состояние системы описывается вектором в -мерном гильбертовом пространстве, что позволяет обрабатывать экспоненциально больше информации, чем классические системы.
Квантовая химия использует принципы квантовых вычислений для непосредственного моделирования поведения электронов в молекулах. В отличие от классических методов, требующих приближений для решения уравнения, квантовые алгоритмы, такие как (Variational Quantum Eigensolver) и (Quantum Phase Estimation), позволяют напрямую рассчитывать энергию основного состояния и другие свойства молекул. В определенных случаях, особенно при моделировании сильно коррелированных систем, это обеспечивает экспоненциальное ускорение по сравнению с классическими подходами, что делает возможным расчет свойств молекул, недоступных для классического моделирования из-за вычислительных ограничений. Экспоненциальное ускорение достигается благодаря возможности квантовых компьютеров эффективно представлять и манипулировать многочастичными волновыми функциями.
Реализация потенциала квантовых вычислений в моделировании молекул требует разработки эффективных алгоритмов, адаптированных к специфике квантовых систем. Ключевой проблемой является необходимость минимизации ошибок, возникающих из-за декогеренции и несовершенства квантовых битов (кубитов). Достижение достаточного количества кубитов с высокой степенью когерентности и точностью управления — сложная инженерная задача, требующая инновационных подходов к материаловедению, криогенной технике и электронному управлению. Кроме того, разработка алгоритмов, способных эффективно использовать ограниченные ресурсы квантового оборудования и масштабироваться для решения задач реального мира, является критически важной для практического применения квантовых симуляторов.
Традиционные методы молекулярного моделирования, такие как методы Хартри-Фока и теория функционала плотности, опираются на приближения для решения уравнения из-за вычислительной сложности. Переход к квантовым вычислениям позволяет напрямую решать это уравнение, избегая необходимости в этих упрощениях. Это достигается за счет использования квантовых алгоритмов, способных эффективно представлять и манипулировать квантовыми состояниями молекул. Вместо получения приближенного решения, квантовые вычисления стремятся к получению точного решения, описывающего поведение электронов в молекуле, что потенциально открывает возможности для моделирования сложных химических процессов и открытия новых материалов с заданными свойствами.
Соединяя Теорию и Практику: Вычислительные Методы
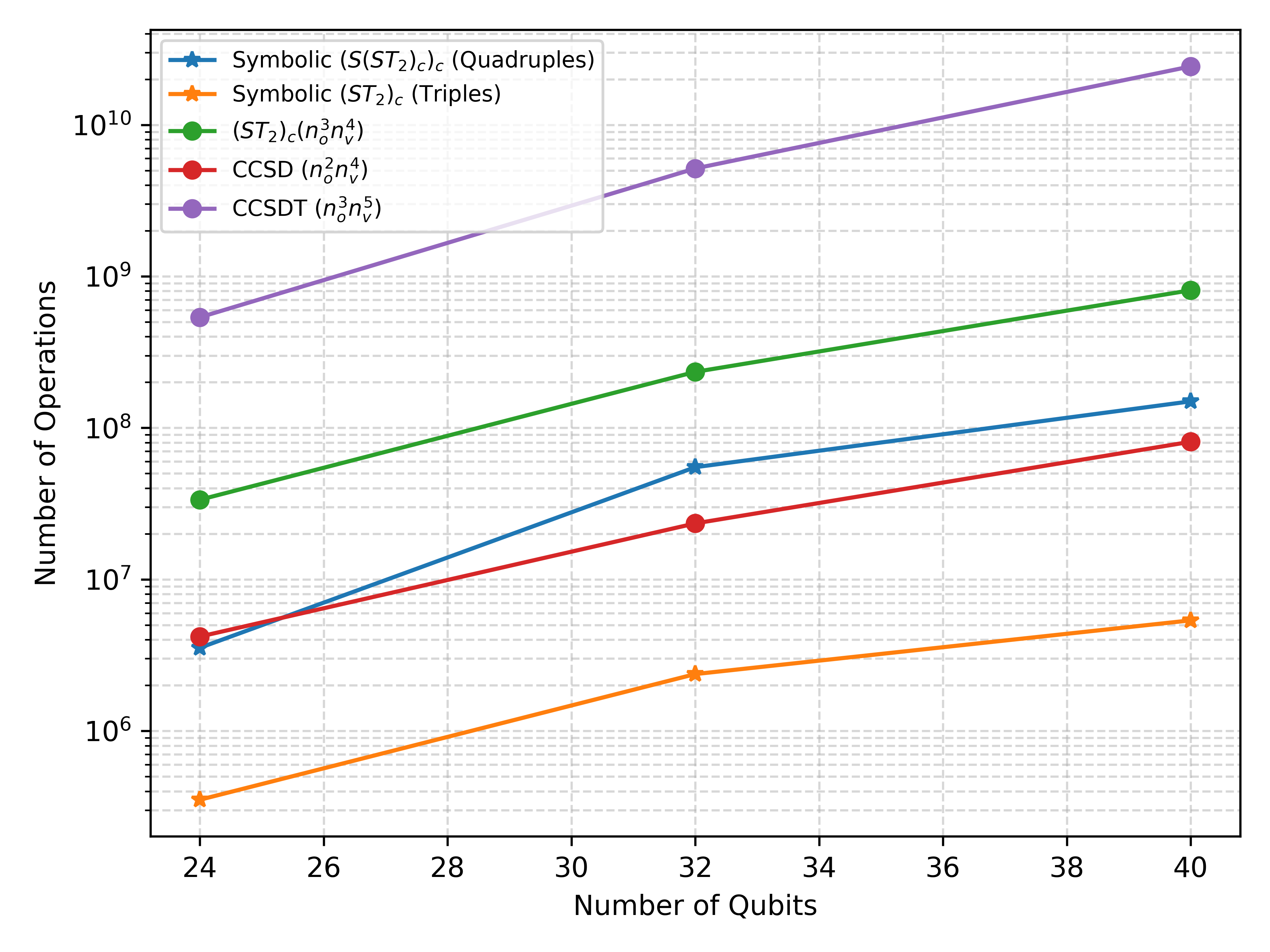
Теория возмущений остается фундаментальным методом в квантовой химии, обеспечивающим систематический подход к приближенному решению уравнения Шредингера. Однако, эффективность этого метода напрямую зависит от скорости и точности вычисления молекулярных интегралов. Эти интегралы, представляющие собой математическое описание взаимодействия электронов и ядер в молекуле, необходимы для расчета энергии и других свойств молекулярных систем. Сложность вычисления молекулярных интегралов возрастает экспоненциально с увеличением числа базисных функций, используемых для описания электронной структуры, что делает их вычисление узким местом в большинстве квантово-химических расчетов. Поэтому, разработка эффективных алгоритмов и методов для вычисления молекулярных интегралов является ключевой задачей для продвижения исследований в области квантовой химии и материаловедения.
Вычисление молекулярных интегралов, необходимых для определения энергий и свойств молекул, часто является основным вычислительным ограничением в квантово-химических расчетах. Сложность этих интегралов растет экспоненциально с увеличением числа базисных функций и атомов в молекуле, что приводит к значительным затратам времени и вычислительных ресурсов. В частности, при использовании методов, требующих точного расчета электронных корреляций, таких как теория возмущений или методы коррелированных кластеров, вычисление и обработка интегралов превосходит по объему все остальные этапы расчета. Оптимизация методов расчета интегралов, включая использование эффективных алгоритмов и параллельных вычислений, является критически важной задачей для расширения применимости квантово-химических методов к более крупным и сложным системам.
Вычислительные методы, основанные на расчете молекулярных интегралов и возмущениях, позволяют проводить исследования сложных систем, таких как FeS-кластеры. Эти кластеры играют ключевую роль в биологической фиксации азота, являясь активными центрами ферментов, катализирующих превращение атмосферного азота в аммиак. Детальное понимание электронной структуры и реакционной способности FeS-кластеров имеет решающее значение для оптимизации процессов фиксации азота, что имеет важное значение для сельского хозяйства и устойчивого развития. Моделирование этих кластеров требует значительных вычислительных ресурсов, но позволяет исследовать механизмы катализа и разрабатывать новые, более эффективные катализаторы.
В настоящее время методы, основанные на вычислении молекулярных интегралов, все шире применяются для исследования высокотемпературных сверхпроводников, расширяя границы материаловедения. Внедрение предложенного рабочего процесса PIGen-SQD позволяет снизить размерность подпространства диагонализации до 70%, сохраняя при этом сравнимую или улучшенную точность по сравнению со стандартным SQD. Это демонстрирует существенное повышение вычислительной эффективности при моделировании электронных свойств данных материалов и открывает возможности для анализа более сложных систем и предсказания новых сверхпроводящих материалов.
Влияние и Перспективы Развития
Точная оценка аффинности связывания лиганда — краеугольный камень современной разработки лекарственных препаратов, позволяющий создавать более эффективные и целенаправленные терапевтические средства. Высокоточные расчеты взаимодействия молекул лиганда с целевыми белками позволяют предсказывать силу и специфичность связывания, что критически важно для оптимизации структуры лекарственного кандидата. Это, в свою очередь, способствует снижению побочных эффектов и повышению эффективности лечения, поскольку позволяет сконцентрировать действие препарата именно на пораженных тканях или молекулярных мишенях. Разработка новых методов, повышающих точность и скорость вычисления — константы диссоциации — открывает перспективы для создания персонализированных лекарств, адаптированных к индивидуальным особенностям организма пациента, и значительно ускоряет процесс вывода новых препаратов на рынок.
Возможность моделирования сложных химических систем открывает беспрецедентные перспективы в разработке новых материалов с заданными свойствами. Благодаря детальному пониманию взаимодействия между атомами и молекулами на квантовом уровне, исследователи получают инструменты для предсказания и оптимизации характеристик материалов ещё до их физического синтеза. Это позволяет целенаправленно создавать вещества с улучшенной прочностью, проводимостью, оптическими свойствами или каталитической активностью, находя применение в самых разных областях — от электроники и энергетики до медицины и машиностроения. Например, моделирование позволяет разрабатывать полимеры с уникальными механическими свойствами или катализаторы, способные эффективно преобразовывать энергию, открывая путь к более экологичным и эффективным технологиям. Такой подход значительно ускоряет процесс разработки материалов, снижая затраты и время, необходимые для создания инновационных продуктов.
Дальнейшее развитие как квантового оборудования, так и вычислительных методов является критически важным для реализации всего потенциала квантовой химии. Успехи в создании более стабильных и масштабируемых кубитов, а также разработка новых алгоритмов, способных эффективно использовать эти ресурсы, позволят моделировать молекулы и материалы с беспрецедентной точностью. Это, в свою очередь, откроет возможности для проектирования новых катализаторов, лекарственных препаратов и материалов с заданными свойствами, недостижимыми при использовании классических методов. Увеличение вычислительной мощности позволит решать задачи, которые в настоящее время недоступны, например, точное моделирование сложных химических реакций и предсказание свойств новых соединений с высокой достоверностью. Таким образом, синергия между аппаратным и программным обеспечением станет ключевым фактором, определяющим будущее квантохимических исследований и их применение в различных областях науки и техники.
Схождение различных научных дисциплин, в частности квантовой химии и материаловедения, обещает фундаментально изменить наше понимание химических процессов и свойств материалов, открывая новые горизонты для инноваций в широком спектре областей. Разработанный PIGen-SQD алгоритм продемонстрировал значительное повышение вычислительной эффективности за счет сокращения пространства поиска до 75%, что позволяет проводить более быстрый и точный анализ сложных химических систем. Это особенно важно при разработке новых лекарственных препаратов и материалов с заданными характеристиками, поскольку позволяет исследовать большее количество потенциальных кандидатов в значительно сокраченные сроки и с меньшими затратами вычислительных ресурсов. В перспективе, дальнейшее развитие этого подхода может привести к созданию принципиально новых технологий и материалов, которые будут востребованы в различных отраслях промышленности и науки.

Исследование демонстрирует, что стремление к ускорению квантово-центричных вычислений требует не просто наращивания вычислительных мощностей, но и переосмысления самой парадигмы вычислений. Алгоритм PIGen-SQD, объединяющий физически обоснованное машинное обучение с квантовой инфраструктурой, представляет собой попытку вырастить, а не построить решение для сложной задачи расчета электронной структуры. Как заметил Нильс Бор: «Противоположности противоположны, но не исключают друг друга». Это особенно верно для гибридных алгоритмов, где классические и квантовые подходы дополняют друг друга, позволяя обойти ограничения каждого из них и достичь большей эффективности в рамках конфигурационного взаимодействия. По сути, это не контроль над вычислениями, а создание экосистемы, способной к самокоррекции и адаптации к будущим сбоям.
Куда же дальше?
Представленная работа, подобно семени, упавшему в плодородную почву, скорее указывает направление, нежели завершает путь. Алгоритм PIGen-SQD, стремясь к ускорению квантово-центрических вычислений, обнажает фундаментальную дилемму: каждая оптимизация — это пророчество о будущей точке отказа. Уменьшая стоимость восстановления конфигураций, он лишь отодвигает проблему к следующему, неизбежно возникающему, узкому месту. Система не становится проще, она лишь меняет форму своей сложности.
Основным вызовом остается не столько скорость вычислений, сколько сама природа приближений, используемых в квантовой химии. Невозможно построить идеальную модель; можно лишь научиться выращивать сад, где ошибки прощаются и компенсируются. Будущие исследования должны быть направлены не на устранение погрешностей, а на создание механизмов, позволяющих системе адаптироваться к ним и извлекать пользу из кажущегося хаоса.
В конечном счете, истинный прогресс заключается не в создании более мощных инструментов, а в развитии более глубокого понимания самой реальности, которую эти инструменты призваны исследовать. Иначе говоря, задача не в том, чтобы быстрее решать старые уравнения, а в том, чтобы задавать новые, более осмысленные вопросы.
Оригинал статьи: https://arxiv.org/pdf/2512.06858.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Статья также опубликована на личном сайте автора.